very hard protein modeling tests|protein model accuracy calculator : company Local quality is valuable for evaluation of local structure error and model refinement, while global quality is valuable for model ranking and selection. This article . Boomerang.bet e direitos relacionados pertencem ao Casbit Group N.V. incorporado em Curaçao, Zuikertuintjeweg Z/N (Zuikertuin Tower), pelo registro digital. número 157923. Confie novamente em OU com número de registro 16310234 e endereço Harju maakond. Tallin, Kesklinna linnoasa é um agente de pagamento.
{plog:ftitle_list}
Agência 2472 Cursino - Caixa Econômica Federal. Confira o .
In a protein structure prediction process, the estimation of model accuracy (EMA) or model quality assessment (QA) without the knowledge of native/true structures is important for selecting.
Local quality is valuable for evaluation of local structure error and model refinement, while global quality is valuable for model ranking and selection. This article .
To investigate how residue-residue distance/contact features may improve protein model quality assessment with deep learning, we developed several EMA predictors to . Tested on the CASP12 and CASP13 datasets, our experimental results show that our method greatly outperforms existing state-of-the-art methods. Our ablation studies indicate .The single model method extracts the sequence, geometric structure, and physical and chemical features of a single protein model, and inputs it into the neural network to predict the quality . Template-free protein structure prediction remains an important unsolved problem. We proposed a new pipeline to construct full-length protein structures by coupling multiple-level deep learning potentials with fast gradient .
protein model accuracy chart
Our main aim with this resource is to provide a standardized way to evaluate pre-trained protein models, and to provide clearer and more informative comparison between .
We carried out a comparative analysis of several popular model assessment methods (RMSD, TM-score, GDT, QCS, CAD-score, LDDT, SphereGrinder and RPF) to . Nature Communications - The use of protein language model embeddings has demonstrated state-of-the-art performance in many protein prediction tasks. Here, authors .
In this work, we propose a novel approach for protein model quality estimation using protein structural stability information obtained from MD simulations. We introduce three features: root-mean-square deviation (rmsd), . Self-supervised language modeling is a rapidly developing approach for the analysis of protein sequence data. However, work in this area is heterogeneous and diverse, making comparison of models .
protein model accuracy calculator
With AlphaFold2 (AF2) has made significant breakthroughs in predicting protein tertiary structures and self-assessment, researchers have shifted their attention from monomers to protein complexes [27], [51], [52].Currently, the accuracy of protein complex modeling is much lower than that of monomer modeling [7].Complex EMA methods are critical to improve their .
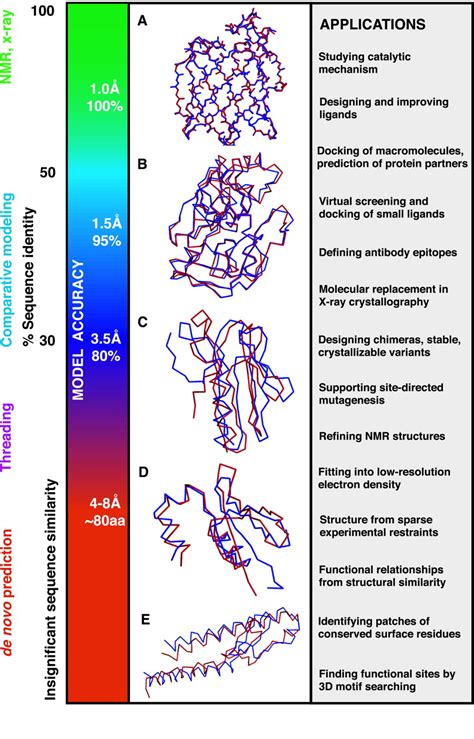
Even very long missing fragments in protein structures could be effectively modeled. Resolution of such models is usually on the level 2-6 Å, which could be sufficient for guiding protein engineering. . including the hard . The estimation of protein model quality remains a challenging task and is important for protein structural model utilization. In the last decade, existing methods that rely on machine learning to deep learning have been developed and shown progressive improvement. Despite utilizing more sophisticated techniques and introducing new features, none of these methods . The three-dimensional modeling of protein structure is a reliable approach for understanding the biochemical functions along with the dynamics of protein interactions, which provides useful applications in developing drug molecules for curing diseases as well as certain other applications in biological and agricultural sciences. There are two sets of criteria that should be considered in an evaluation of full-atom refined models. The first set is based on the global topological similarity of the model to the experimental structure, including the root mean-square deviation (RMSD) (5), template modeling (TM)-score (6), and global distance test-total score (GDT-TS) (7).The second set is based on .
lddt protein model
Proteins are molecular devices, in the nanometer scale, where biological function is exerted (1). They are the building blocks of all cells in our bodies and in all living creatures of all kingdoms. Although the information necessary for life to go on is encoded by the DNA molecule, the dynamic process of life maintenance, replication, defense and reproduction are carried out .The levels of protein structure organization (PDB code 1AXC). (a) A sequence is the primary structure of the protein. The three-dimensional structure of the protein consists of several regular secondary structures such as helices (b) and β-sheets (c).(d) The secondary structures are linked by loops (colored black) and form a tertiary structure.(e) Multiple folded proteins can be .
General-purpose foundation models are very useful when data are abundant because they’re able to capture the full generality of a problem, as AlphaFold2 did with protein structure prediction .From fold recognition to homology modeling: an analysis of protein modeling challenges at different levels of prediction complexity . far. The RMSD of C-alpha atoms are 1.2-1.7, 2.3, and 4.6-17.9 A for the three easy targets, two hard targets, and three very hard homology targets, respectively. Out of 18-fold recognition predictions available .
AlphaFold predicts protein structures with an accuracy competitive with experimental structures in the majority of cases using a novel deep learning architecture.
Protein Modeling is a Division C event for the 2021 season. It was introduced as a trial event in 2009 and as an official national event in the 2010-2011 season, 2011-2012 season, 2014-2015 season, 2015-2016 season, 2018-2019 season, and the 2019-2020 season.For the event, students use computer visualization and online resources to guide the construction of .In Go (the game with black and white stones), the state space is fairly small: 3 361, since each of 19*19 locations can be either unoccupied, black, or white.In other words, there are 361 slots of 1 degree of freedom, where each slot has 3 possible values. In the 1950s, Anfinsen suggested that the amino acid sequence of a protein encodes the information required to guide a protein folding event. 1 The result of the protein folding experiments was the achievement of a defined structure or shape of a protein capable of carrying out its specific function. This crucial understanding allowed the researchers to study . Further, protein modeling has advanced rapidly over 50 years, even slightly faster than Moore's law. We also review an interesting scientific social construct that has arisen around protein modeling: community-wide .
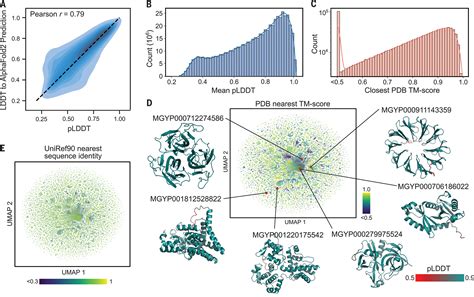
Here the authors build deep learning predictors of protein expression from sequence that deliver accurate models with fewer data than previously assumed, helping to lower costs of model-driven . The identification of protein homologs in large databases using conventional methods, such as protein sequence comparison, often misses remote homologs. Here, we offer an ultrafast, highly . On average, the pLDDT of DMFold models (0.663) is 11% higher than that of AlphaFold2 DB models (P < 2.2 × 10 −16, one-sided Student’s t-test), and 94% (4,738 of 5,042) of the DMFold models .At present, there are about 87,000 structures in the Protein databank, and these span about 1393 folds, so that a homology model can be produced for in excess of half of all protein domains of known sequence. 4,5 Homology models vary greatly in accuracy depending on a number of factors, and for that reason CASP has encouraged the development of .
This requires large amounts of very pure protein and often involves years of trial and error, searching for the proper crystallization conditions. . The reliability of the final atomic model depends on the resolution of the original crystallographic data: 0.5 nm resolution might produce a low-resolution map of the polypeptide backbone . To predict the in vivo PD response, the predicted protein degradation (Eq.3) is integrated (Eq. 7) with the TPD's inhibitory effects as calculated from the concentration–time profile (Eq. 8).The method applied to derive prediction intervals is analogous to the one used for in vivo degradation.. Software implementation. The model was built and validated with in-house .
The estimation of model accuracy (EMA) category has been an integral part of every CASP experiment starting with CASP7. 41-48 It has attracted the attention of many developers, with over 70 methods tested in the previous CASP experiment. 48 An emphasis on the importance of this category led to very positive developments in protein structure .
De novo design provides a rigorous test of our understanding of protein structure and allows the . acid sequence for a protein can be very sensitive to the precise 3D structure of the protein . From the MODELLER web site. MODELLER is used for homology or comparative modeling of protein three-dimensional structures (b and Sali 2016, Marti-Renom et al. (2000)). The user provides an alignment of a sequence to be modeled with known related structures and MODELLER automatically calculates a model containing all non-hydrogen atoms. While recent advances in modeling protein function in response to mutations have focused on increasing the capacity of models to fit more complex fitness landscapes 5,6,7,8,9,10, it remains .
Protein modeling is an important method for understanding the structural basis of a protein. The structural information derived from a modeled protein along with the corresponding pharmacophores can be applied to predict binding potentials of candidate binders and to rationalize molecular structure–activity relationship for drug design. Fig 4g illustrates a high-quality model for a very large protein complex . the sequence match score and the overall amino acid confidence type score for the 167 test structural models is 0.7, .
web3 de jun. de 2023 · Villarreal x Atlético de Madrid: onde assistir, horário e escalações do jogo pela La Liga - Lance! Resenha de Apostas. Página Principal. Dicas de Apostas. .
very hard protein modeling tests|protein model accuracy calculator